基于三代测序技术的微生物组学研究进展 _sanger et al.,1978-程序员宅基地
基于三代测序技术的微生物组学研究进展
2020-09-04 09:16
微生物通常指一切难以用肉眼观察到的微小生物, 包括细菌、病毒、古菌、真菌以及一些微小的原生生物。微生物体积微小、结构简单, 却又无处不在, 在人类健康(Yang & Tarng, 2018)、工农业生产(Huang et al, 2018)、环境保护(Liu et al, 2018)和食品安全(Alori & Babalola, 2018)等方面都发挥着重要的作用。微生物传统的研究方法以分离纯菌培养为主, 但由于培养技术的限制, 超过99%的原核微生物无法在实验室培养得到, 导致微生物结构和功能多样性的研究一直发展缓慢(Schloss & Handelsman, 2005)。
16S/18S rRNA基因存在于所有微生物的基因组中, 是微生物分类鉴定最常用的标记分子。Sanger测序技术的兴起(Sanger et al, 1977), 使得16S rRNA基因测序在细菌分类学中被广泛应用, 由此发现了大量新的微生物门类, 增加了微生物的多样性。1998年提出的宏基因组学(Hugenholtz et al, 1998)方法, 是直接从环境样品中获取总的遗传物质来研究微生物群落的组成和结构, 发现新的功能基因。宏基因组学的方法不仅解决了微生物分离培养的难题, 而且可以全面分析微生物群落的多样性和丰度, 研究微生物之间、微生物和环境或宿主之间的关系。而在过去的数年间, 第三代测序技术的出现, 以其超长的测序读长、无GC碱基偏好性和速度快等优势, 在以16S/18S rRNA基因或全基因组为目标序列的微生物组学研究中取得了一系列进展, 因此微生物的组成和群落结构、代谢特征、系统进化、微生物与环境的相互作用得到了更深层次的研究。
1 测序技术的发展
1.1 第一代测序技术
第一代测序技术(Sanger法)开始于1977年, 该测序方法完成了噬菌体X174全长5,375个碱基的基因测序, 由此生命科学研究进入了基因组学时代(Sanger et al, 1977)。Sanger法即双脱氧终止法(Chain Termination Method), 是指在含有四种脱氧核苷三磷酸(dNTP)的反应体系中加入一种不同的荧光标记的双脱氧核苷三磷酸(ddNTP)。由于ddNTP没有DNA延长所需要的3-OH基团, DNA合成随机地在G、A、T、C处终止, 然后通过凝胶电泳确定待测DNA分子的长度。第一代测序技术的测序读长在1,000-1,500 bp, 但一次只能测一条单独的序列, 导致测序成本高、通量低, 不能够应用于大规模的基因测序。
1.2 第二代测序技术
二代测序技术又称“下一代”测序技术(Next-Generation Sequencing Technology, NGS)。该技术在保持测序准确度的前提下, 主要解决了第一代测序通量低的问题。二代测序技术可以同时对几万到几百万条DNA分子进行测定, 因此也被称为高通量测序技术(Metzker, 2010)。二代测序平台主要有Roche公司的454 (Sogin et al, 2006)、Illumina公司的Solexa、Hiseq (Gloor et al, 2010)和ABI公司的Solid等测序仪(Mardis, 2008), 其原理分别为焦磷酸测序、边合成边测序、连接测序。二代测序技术既大大降低了测序成本又大幅提高了测序通量, 同时保持了高准确性。比如使用Illumina公司的技术平台, 在30×测序深度下, 完成人类基因组测序仅仅需要1周(Niedringhaus et al, 2011)。但二代测序技术的主要问题在于测序读长短, 只有250-300 bp, 而且由于测序的系统偏好性, 导致有些序列可能被测了很多次, 但也有一些量少的序列无法被大量扩增, 造成信息的丢失。
1.3 第三代测序技术
第三代测序技术主要有Pacific Biosciences公司的SMRT (Single Molecule, Real-Time Sequencing)技术和Oxford Nanopore公司的Nanopore技术(Eid et al, 2009)。三代测序技术的主要特点是单分子实时测序、长读长, 但错误率偏高。下面从技术特点和测序原理两方面来介绍第三代测序技术。
1.3.1 SMRT技术
SMRT技术的核心是能够实现单个DNA分子的测序, 并且实时监控测序结果。2013年Pacific Biosciences公司成功推出商业化的三代测序仪PacBio RSII后, 三代测序开始被广泛应用于微生物基因组研究中。经过不断的改良和升级, 又在2015年10月推出全新的PacBio Sequel测序系统, 测序技术的准确性和通量不断提高。SMRT技术测序系统主要有SMRT Cell、零模波导孔(Zero-Mode Waveguides, ZMW)和DNA聚合酶。
SMRT Cell是测序芯片, DNA样本完成建库后装载到Cell中才能上机测序。PacBio RSII系统配套的一个SMRT Cell的碱基产量为500 M到1 G, 而最新升级的Sequel系统配套的一个SMRT Cell可产生5-10 G的数据, 测序通量提升10倍左右。零模波导孔ZMW是一种直径为50-100 nm的圆形纳米小孔。当DNA分子进入小孔后, 因为从孔底发出的激发光不能穿透小孔进入上方的溶液区, 被限制在底部一个足以覆盖被检测DNA部分的区域, 而共聚焦显微镜只收集该区域的信号, 从而将背景噪音降到最低。PacBio RSII使用的一个SMRT Cell中含有15万个ZMW孔, 而最新的Sequel平台配套的升级版SMRT Cell中含有100万个ZMW孔。DNA聚合酶是实现SMRT技术长测序读长的关键之一, 它在保持测序延续性的基础上, 实现了DNA体内的合成速度。DNA聚合酶能够每秒钟读取10个碱基, 因此测序速度是化学测序方法的2万倍。
1.3.2 Nanopore技术
与以往的测序技术均不同, Nanopore是采用电信号进行测序(Magi et al, 2017)。2014年, Oxford Nanopore公司开始对外提供Nanopore MinION试用项目计划, 随后不断对早期版本仪器的高错误率和低通量问题进行改善。从2016年开始, Nanopore平台的通量得到较大提升, 错误率也显著降低, 在基因组中的应用也从微生物小基因组逐渐延伸到复杂动植物基因组。而更高通量测序平台GridION X5 和PromethION的发布使Nanopore在复杂物种中的应用更为简单和便捷。新的测序仪在延续MinION核心测序技术的操作简单和文库制备快等优点的基础上, 弥补了MinION测序仪通量低以及不适用于大批量样本或大基因组测序的不足。根据最新的文献记载, Nanopore MinION测序基于新开发的BulkVis工具首次生成了单个2.2 M序列(Payne et al, 2018)。
Nanopore测序系统主要包括纳米孔、薄膜和马达蛋白(motor protein)。纳米孔是一种跨膜蛋白形成的纳米孔道。不同版本的测序芯片使用不同的跨膜蛋白, 目前R9版的芯片使用的是来自大肠杆菌的CsgG蛋白质经过基因工程改造后形成的通道。薄膜是人工合成的具有高电阻的膜, 膜的两侧浸在含有离子的水溶液中, 通过对膜上施加电势, 离子通过薄膜上的蛋白纳米孔产生电流。马达蛋白是一种DNA解旋酶, 在构建文库时, 马达蛋白和引导接头(leading adaptor)一同加在DNA分子上, 在测序过程中, 马达蛋白会对双链DNA解螺旋使其变为单链, 使得单链DNA以一定速度经过纳米孔。
Nanopore技术有3种不同的建库方式: (1)在1D 建库中, 仅有引导接头, 在测序过程中, 马达蛋白对双链DNA解压和解链, 引导接头通过纳米孔, 随后模板链通过。(2)在2D建库中, 既有引导接头, 还有连接双链DNA分子的发夹接头(hairpin adaptor)。
在测序过程中, 首先是马达蛋白对双链DNA解螺旋使其变为单链, 引导接头通过纳米孔, 随后模板链通过, 然后发卡接头和互补链通过。(3)在1D2建库中, DNA双链分别通过纳米孔, 但并未如2D测序中通过发卡接头进行连接。当模板链完成测序后, 纳米孔会捕获互补链的马达蛋白进行互补链测序(Clarke et al, 2009)。1D测序建库的优势在于文库构建更便捷, 可低至10 min; 相对1D测序, 2D测序中可以得到更长的读长; 1D2同时对模板链和互补链进行测序, 可以得到高质量的一致性序列(Jain et al, 2018)。
2 三代测序技术在微生物组学研究中的应用
2.1 16S/18S三代测序
16S rRNA基因存在于所有原核微生物的基因组中, 序列长度约为1,500 bp (18S rRNA基因存在于所有真核微生物的基因组中, 序列全长为1,500-2,000 bp)。16S rRNA基因有9个高变区(V1-V9), 在一定程度上反映了微生物间的进化差异, 可以利用高变区对不同菌属、菌种的细菌进行分类鉴定; 而与高变区间隔分布的8个高度保守区域在细菌漫长的进化过程中则保持了结构和功能的稳定性。1977年, Woese等依据细菌16S rRNA基因序列上的区别, 提出了三域系统, 并分别命名为细菌域、古菌域和真核域(Woese & Fox, 1977)。目前16S/18S rRNA基因是研究微生物系统发育和分类鉴定最常用的分子标记(Woese, 1987)。
基于16S/18S的微生物基因测序即是利用16S/18S rRNA保守区域的基因序列设计引物, 扩增研究所需的基因片段(Jonasson & Monstein, 2002)。包括基因组DNA的获取; 设计标准引物对16S/18S基因的一个或多个区进行扩增; 聚类, 按照一定的相似性将扩增得到的序列进行分组, 细菌中的分类操作单元(operational taxonomic unit, OTU)是基于序列97%相似性的分类; 然后通过数据库比对为OTU的代表性序列提供分类学注释。
图1
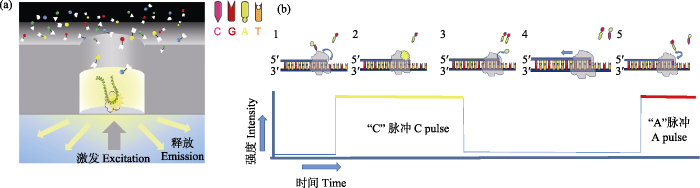
图1 PacBio SMRT测序原理。(a)在零模波导孔(Zero-Mode Waveguides, ZMW)中, 单个DNA分子模板与引物和聚合酶结合后, 被固定到ZMW孔底部。DNA合成开始时, 新加入的荧光标记的dNTP由于碱基配对在ZMW底部停留较长时间, 激发后发出对应的荧光信号被共聚焦显微镜实时记录; (b) 1)荧光标记胞嘧啶脱氧核苷酸; 2)胞嘧啶脱氧核苷酸进入DNA链配对, 发射荧光信号; 3)荧光基团被DNA聚合酶切除, 荧光消失; 4)标记新的脱氧核苷酸; 5)继续新一轮合成。
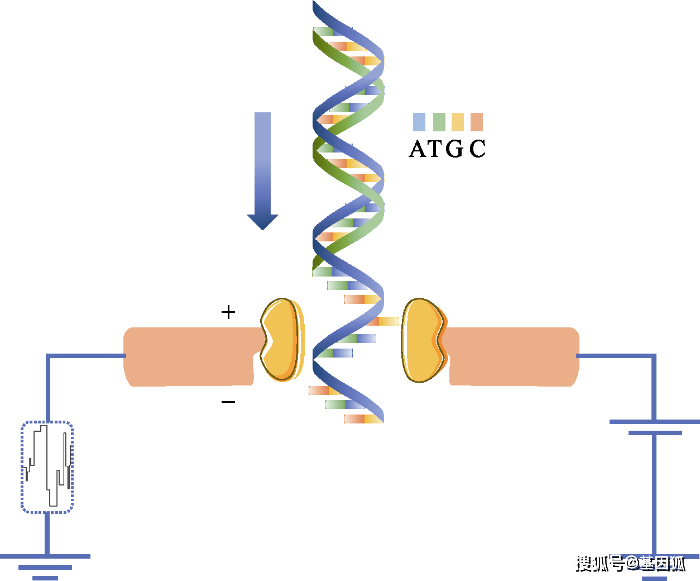
图2 Nanopore利用电信号检测出DNA的碱基序列。纳米孔直径很小, 仅仅允许单个核苷酸通过。当DNA单链通过的时候, 就会对离子的流动造成阻碍, 从而使流过纳米孔的电流强度发生变化。由于ATCG四种碱基的带电性质不一样, 造成电流大小的波动也不一样, 因此可根据电流的变化鉴定所通过的碱基类型。
三代测序技术由于超长的测序读长, 在微生物的分类鉴定和群落多样性的研究中更具有优势。二代测序通常只能获得1-2段高变区域, 而三代测序的读长提升了2-3倍以上, 因而也提升了物种鉴定的分辨率。研究人员对同一人的肠道菌群样本分别进行二代和三代的16S rRNA基因测序, 其中使用Illumina Miseq平台对16S rRNA基因的V3-V4区进行扩增测序, 使用PacBio SMRT技术直接读取和覆盖16S rRNA基因的V3-V7区, 实验结果表明有更多的OTUs基于SMRT测序数据被鉴定出来(Franzen et al, 2015)。Cusco等(2018)将Nanopore MinION应用于16S rRNA基因测序, 在犬类的皮肤微生物群体中发现了新的细菌门类。在另外一项针对已知菌种混合组成的模拟菌群的研究中, Nanopore MinION的测序数据使得一些物种能鉴定到种的水平(Benitez-Paez et al, 2016)。因此当三代测序技术的读长可以覆盖到更多基因片段时, 测序结果能够更准确地检测到微生物群体多样性组成。
2.2 细菌/真菌基因组三代测序
在微生物组学的研究中, 已有参考基因组的微生物数量远远低于自然界存在的微生物数量, 除了微生物难以纯化培养外, 还有一部分原因是二代测序读长短, 难以解决细菌/真菌基因组中的高重复区域和高GC区域的组装问题, 因此组装后的基因组中往往存在很多gap。而三代测序技术以其超长的测序读长和无GC偏好性克服了以上部分难题, 在单细菌/真菌的基因组组装中取得了很大突破。
2013年Chin等就使用SMRT技术组装出16个微生物基因组, 与参考基因组一致性达99.9999% (Chin et al, 2013)。Brown等(2014)分别使用Illumina和454这些二代测序平台和PacBio RS II平台技术对梭状芽孢杆菌(Clostridium autoethanogenum)进行基因组装, 发现二代测序平台组装得到Clostridium的基因组仍然存在一些gap, 而使用PacBio SMRT测序直接组装得到一条完整的Contig; 并且之前由于技术的限制, 无法区分C. autoethanogenum和C. ljungdahlii两支菌株, 而基于PacBio SMRT的全基因组测序, 发现两者在CRISPR系统、氢化酶等方面具有显著差异。Ludden等(2017)用Nanopore MinION测序平台首次组装出了Enterobacter kobei的基因组完成图, 并且组装出编码blaOXA-48基因的质粒。Wick等(2017)对12个不同种的克雷伯氏肺炎菌进行测序, 然后与前期二代数据混合组装, 既保证了测序数据的完整性, 又保证了测序的准确性, 最终都组装成了基因组完成图。此外假单胞菌Pseudomonas koreensis P19E3的基因组含有长达70 kb的重复序列, 使用二代平台的测序数据无法组装到完整的基因组, 而Schmid (2018)使用Nanopore MinION 测序仪, 首次完成了对Pseudomonas koreensis P19E3的基因组组装。
真菌基因组大小在2.5-150 Mb之间, 介于细菌和大型动植物之间, 随着三代测序平台的改进和升级, 测序通量的不断提升, 三代测序技术在基因组研究中的应用也逐渐从细菌等小型基因组延伸到真菌等大型基因组中。Orpinomyces sp. strain C1A菌株是一种寄生在大型家畜肠胃中的厌氧真菌, 基因组超过100 Mb, GC含量只有17%, 是目前已知的GC含量最低的物种之一。Yousse等(2013)在对该菌株的基因组组装中发现, 对比Illumina测序数据得到的组装结果, PacBio RS的测序数据组装得到的contigs更长、数量大幅降低, 而且发现C1A菌株具有降解木质素的能力。此外Faino等(2015)基于PacBio RS II平台对黄萎病菌菌株JR2和VdLs17测序并组装, 最终都得到了8条完整的染色体。2017年荷兰研究者Jansen等分离出了DDNA#1酵母菌株DNA, 使用Illumina和Nanopore MinION分别进行测序, 通过Illumina测序数据组装得到了14,764 contigs, 而使用Nanopore测序数据组装得到的contigs仅61条, 大幅度提高了酵母菌基因组数据的质量和完整度(Liem et al, 2017)。
表1 三代测序技术的比较
2.3 宏基因组三代测序
宏基因组学是将环境中的微生物群落作为整体进行研究, 与16S/18S或全基因组分析相比, 宏基因学能够对微生物群体基因组成及其功能、微生物群体的多样性、微生物与环境、微生物与宿主之间的关系进行全面解读。2016年Hug等就利用宏基因组技术发现了1,000多种未被培养或者了解甚少的微生物体, 进一步扩展了微生物的多样性, 而新的细菌门的出现, 也重新补充了进化树三域系统(Hug et al, 2016)。Fierer等(2007)通过构建牧场、沙漠、雨林土壤宏基因组文库, 对环境中细菌、古生菌、真菌及病毒多样性进行了研究。结果发现土壤环境中包含着大量不可培养的新病毒种类, 其基因型特征与常规培养获得的病毒具有很大的差异。除此之外, 宏基因组学在海洋微生物多样性的探索中也取得了很大的进展, 促进了海洋微生物资源的开发。2015年Sunagawa领导的团队分析了来自世界各地的68个地点的远洋中上层水域的共243个样本, 构建了含有4 × 107个非冗余基因的海洋微生物宏基因组库, 并提出在垂直水平上海洋微生物群落的变化主要是由温度决定的; 同时对139个富含原核微生物的样本进行统计分析, 共发现了5,755个同源微生物组, 其中有40%的同源组与人类肠道菌群的同源组相同(Sunagawa et al, 2015)。
二代测序应用于宏基因组学时, 由于读长过短会导致一些基因信息的丢失。相比之下, 宏基因组学三代测序结果可以更加真实地反映菌群的组成情况, 较为准确地挖掘出新的功能基因。例如研究人员使用二代和三代测序平台分别对同一微生物发酵池样品进行了宏基因组测序分析, 并对结果做了比较。发现基于Illumina测序数据的组装, 由于读长较短, 结果存在大量不同物种间的同源序列错误组装, 无法真实呈现菌群组成; 而PacBio RS II得到的结果真实反映了两种优势菌株的组成情况(Frank et al, 2016)。Tsai等(2016)也采用了PacBio RS II和Illumina HiSeq两种测序平台对人类手部和足部的菌群进行宏基因组分析, 结果表明三代测序技术显著减少了contigs的数量, 大大降低了序列拼接和基因组组装的难度, 而且从人的皮肤菌群样本中组装、注释、构建获得一例未知微生物的高质量基因组。在对Sakinaw Lake水体微生物的研究中, Singer等(2016)发现SMRT和Illumina的测序数据在门的水平上没有太大差别, 但是随着群落复杂度的增加, 二代和三代的测序数据在群落结构和系统发育分辨率上显示出显著差异。
新型三代测序设备的出现还扩展了宏基因组研究的应用场景。目前基因测序的工作主要局限于实验室, 而Nanopore MinION这种便携式测序仪为开展基因组实时测序提供了便利, 在鉴定新的病原体和细菌基因组测序方面得到了广泛应用。例如猪的肠道疾病通常是由不同的病原体引起的, 宏基因组学是一种常用的诊断方法, 而使用Nanopore技术在3小时内就可以完成样品测序和致病菌的诊断(Theuns et al, 2018)。MinION测序仪对于样本的预处理要求也较低, 对一些高致病性病毒如奇昆古尼亚病毒、埃博拉病毒和丙型乙肝病毒, MinION测序仪可以直接使用临床样本进行检测, 而不需要病毒的纯化培养(Greninger et al, 2015)。在埃博拉等暴发疾病的实时检测中, 研究人员就利用这种便携式测序仪在整个基因组库中鉴别出了新型的单核苷酸多态性(SNPs), 为流行病暴发的预警和病原体的进化提供了重要的方法(Quick et al, 2016)。
目前RNA病毒的基因组研究通常是先反转录为cDNA再进行测序。最近美国的研究者通过设计一种针对流感病毒基因组的Nanopore测序接头RTA, 实现了对流感病毒RNA的直接测序(Callaway, 2018)。而且传统方法对表观遗传修饰的分析, 需要对样本进行亚硫酸盐处理或者抗体沉降(pull-down)的预处理, 增加了检测的难度。通过Nanopore技术, Andrew Smith研究团队直接对大肠杆菌全长16S核糖体RNA进行测序, 从采样到出结果只需要2小时。通过数据分析, 该研究小组在16S rRNA的已知位点鉴定出了7-甲基鸟苷(m7G), 并且观察到了假尿嘧啶核苷等在内的表观遗传修饰的存在(Smith et al, 2017)。中国科学院水生生物研究所赵亮博士通过PacBio RS II测序, 获得3个微囊藻菌株的全基因组甲基化修饰图谱, 发现了18个未被鉴定的甲基化模块, 也发现不同微囊藻菌株的甲基化和甲基转移酶含量差异很大(Zhao, 2018)。
三代测序技术的兴起, 极大地促进了微生物组学的发展, 但是错误率偏高是三代测序技术面临的最大问题。15%-40%的错误率, 大大地高于二代测序技术(< 1%), 成为限制其商业应用开展的重要原因。不过三代测序的错误是随机发生的, 可以靠覆盖度来纠错(但这又要增加测序成本)。为了提升准确率, PacBio公司对SMRT技术配套的技术不断优化, 文献中经常看到的P4C2、P5C3以及P6C4代表的就是不断升级的试剂。同时, PacBio独有的环形一致性测序模式(circular-consensus sequence, CCS)极大地提高了单碱基测序的准确率。Nanopore公司也不断从Flow Cell、纳米孔、测序试剂和信号捕获及碱基识别软件等方面进行升级改进。其中, 纳米孔最开始为R6, 后来不断升级而出现了R7, R8, R9版本, 到现在已经升级为R9.4, 随之而来的是准确率和通量的提升。而且目前已经有比较成熟的软件Canu专门处理三代技术这类错误率较高的测序数据。Canu采用了先纠错、修整再组装的策略, 通过序列与序列之间的重叠, 进行单碱基的错误纠正, 测序覆盖度越高, 准确率就越高。
 三代测序技术的比较
三代测序技术的比较
3 结语
近年来, 微生物组学是生命科学研究的热门领域, 而微生物组的深入研究和理解得益于测序技术的快速发展。第三代测序技术的出现, 实现了很多技术上的突破: (1)凭借其超长读长的优势, 解决了二代测序技术读长过短的问题, 在微生物基因组学研究中减少了后续的基因组组装和注释的工作量, 节省了大量的时间; (2) SMRT技术利用DNA聚合酶自身的合成速度, 配上高分辨率的光学检测系统, 实现了实时快速检测; (3)三代单分子测序技术的发展也让通过测序实时读取DNA碱基修饰成为可能, 这对在基因组水平直接研究表观遗传现象有极大的帮助; (4) Nanopore技术还可以直接对RNA进行测序, 省去了反转录过程。
但由于三代测序较高的错误率, 目前三代测序技术在微生物组中的研究往往需要与二代测序数据结合使用, 即利用二代测序数据对三代测序数据进行校正, 也就意味着在实验中至少要制备2个不同的文库, 造成了一定的人力和物力成本的增加。随着三代技术的不断改进和新的组装方法的出现, 测序的准确性和通量也在逐步上升。相信在不久的将来, 基于三代测序技术的微生物组学研究能够从获得高质量的基因组装结果, 逐渐转向对物种生物学特征和进化历程的深入研究, 研究策略也由单一组学测序逐渐延伸为基因组、转录组、代谢组和表观遗传组的多组学分析。
参考文献(略)
- 许亚昆, 马越, 胡小茜, 王军. (2019) 基于三代测序技术的微生物组学研究进展. 生物多样性, 27(5), 534-542.
智能推荐
用Python 绘制多个同心圆 (Python经典编程案例)_python利用负循环画10个同心圆-程序员宅基地
文章浏览阅读4.1w次,点赞12次,收藏14次。案例:绘制多个同心圆代码如下:import turtlet = turtle.Pen()my_colors = ("red", "green", "yellow", "black")t.width(4)t.speed(1)for i in range(10): # 0 1 2 3 4 t.penup() t.goto(0, -i*10) # 0, -100,-2..._python利用负循环画10个同心圆
pki的java实现书籍_精通PKI网络安全认证技术与编程实现 PDF扫描版[214MB]-程序员宅基地
文章浏览阅读240次。精通PKI网络安全认证技术与编程实现从实战出发,介绍了PKI应用开发过程和细节。《精通PKI网络安全认证技术与编程实现》共32章,分6篇,主要内容包括PKI基础知识、OpenSSL开发、CrytoAPI开发、JavaSecurity开发、电子商务网站应用、PKI技术应用等,涉及C语言、Java语言、JSP、ASP/ASP.NET、PHP等开发语言。为了方便读者深入了解PKI,《精通PKI网络安全认..._api安全技术与实战pdf
c语言字符串函数 小数,C语言modf()函数:求双精度数的小数部分-程序员宅基地
文章浏览阅读644次。函数名: modf头文件:函数原型: double modf(double value, double *iptr);功 能: 求双精度数的小数部分参 数: double value 为要操作的双精度数 ,double *iptr 为要传回整数部分的变量指针返回值: 返回value的小数部分程序例: 分别求出双精度number的小数部分和整数部分,并将结果输出#include#includ..._c语言求一个双精度数的整数部分和小数部分
SYN flood_http存在synflood吗-程序员宅基地
文章浏览阅读288次。最近在学习DDos相关知识,参考一些只是,做了摘要,供自己参考。 参考:http://blog.csdn.net/xlf13872135090/article/details/8059538SYN Flood 攻击原理 利用TCP协议缺陷,发送了大量伪造的TCP连接请求,使得被攻击方资源耗尽,无法及时回应或处理正常的服务请求。一个正常的TCP连接需要三次握手,首先客户端发送一个包含SYN标志的数_http存在synflood吗
测牛学堂:软件测试之数据库操作语句sql的外键查询_sql查询外键id的数据-程序员宅基地
文章浏览阅读775次。我们之前学习的都是针对一个表的操作。如果要进行多个表之间的操作,就要用到外键把他们关联起来。外键的作用:能够让多个表进行关联,使表与表之间有联系,实现共性抽取。如果数据项比较多的情况下,把所有数据都存放在一个表中,如果表太大,影响操作效率。解决办法就是把一个表拆分成多个表,并且用外键去关联。例子:如果要设计一个员工表1)员工表:编号、姓名、年龄、性别、所在分公司、所在部门2)部门表:编号、部门名称、部门经理、主要任务3)公司表:编号、分公司名,地址、电话、法人把公司和部门的数据抽取出来,形成_sql查询外键id的数据
OpenGL学习笔记【4】——创建窗口,给窗口添加渲染颜色_opengl窗口颜色-程序员宅基地
文章浏览阅读803次,点赞14次,收藏14次。章节一讲述了OpenGL在渲染的时候需要一个Context来记录了OpenGL渲染需要的所有信息和状态,可以把上下文理解成一个大的结构体,它里面记录了当前绘制使用的颜色、是否有光照计算以及开启的光源等。不同的操作系统,都有各自的上下文创建方法,最简单的上下文可以通过创建。章节二讲述了一个一个轻量级的图形界面框架,GLFW 的是提供了处理手柄、键盘、鼠标输入章节二还创建了一个空项目章节三讲述了GLAD库是用来管理OpenGL的函数指针的,所以在调用任何OpenGL的函数之前我们需要,从而让我们。_opengl窗口颜色
随便推点
opencv: 使用InRange函数进行阈值操作 Thresholding Operations using inRange_inrange和cv2.threshold一起使用-程序员宅基地
文章浏览阅读1.3k次。目标:使用OpenCV cv::inRange 函数进行基本的 阈值操作, 基于像素值在HSV色度空间的范围进行对象检测理论:前一篇文章中我们学习了如何使用cv::threshold 阈值函数进行阈值操作 本文我们将学习使用 cv::inRange 来进行处理 原理是一样的但是现在我们增加了一个我们所需要的 【像素值的范围】HSV色度空间 HSV colorspaceHSV ..._inrange和cv2.threshold一起使用
瑜伽教学法 | 为什么你说的口令会员没反应?_会员病了无法来上瑜伽课怎么说-程序员宅基地
文章浏览阅读154次。 瑜伽培训课程层出不穷,但市面上都没有教授瑜伽老师们如何“教”的系统培训。瑜伽行业表面看似繁荣,但大多数老师缺失教学的“灵魂”。 为此,心合瑜伽学院王梓涵院长结合多年来积累的经验以及瑜伽老师的痛点,与心合教学团队不断打磨,开创瑜伽培训先河,首创贴合瑜伽老师的『瑜伽教学法』,教学法正是指导瑜伽老师们如何上课的法门! 不少老师们,有时会有这样的问题: “我把正确的口令讲出来了,但是会员好像不听我的口令,并没有按照口令去做,需要我不停地辅助和做示范才能完成...” 一个优秀的老师,总可以_会员病了无法来上瑜伽课怎么说
随机森林sklearn FandomForest,及其调参_随机森林及其调参-程序员宅基地
文章浏览阅读2.2w次,点赞12次,收藏113次。随机森林概述随机森林是集成学习方法bagging类中的翘楚。与集成学习boosting类的GBDT分庭抗礼。bagging类集成学习采用的方法是:用部分数据 or 部分特征 or 多个算法 训练一些模型;然后再组合这些模型,对于分类问题采用投票多数表决,回归问题采用求平均。各个模型训练之间互不影响,天生就适合并行化处理。在如今大数据时代背景下很有诱惑力。 主要效果:重点关注降低方差,..._随机森林及其调参
解决Exception: org.apache.hadoop.io.nativeio.NativeIO$Windows.access0(Ljava/lang/String;I)Z-程序员宅基地
文章浏览阅读1.5k次,点赞2次,收藏2次。解决Exception: org.apache.hadoop.io.nativeio.NativeIO$Windows.access0(Ljava/lang/String;I)Z和Error: JAVA_HOME is incorrectly set.Please update D:\Software\hadoop260\conf\hadoop-env.cmd‘-Xmx512m’ 不是内部或外部命令,也不是可运行的程序或批处理文件。这个错误目前我知道的有以下几种解决办法:一、查看hadoop安装是_org.apache.hadoop.io.nativeio.nativeio$windows.access0(ljava/lang/string;i)z
linux 找不到防火墙设置的,linux怎么样查看防火墙有没开启-程序员宅基地
文章浏览阅读2.6k次。我的是linux系统,那么要怎么样才能查看防火墙有没有开启呢?下面由学习啦小编给你做出详细的linux查看防火墙是否开启方法介绍!希望对你有帮助!linux查看防火墙是否开启方法一:service iptables status可以查看到iptables服务的当前状态。但是即使服务运行了,防火墙也不一定起作用,你还得看防火墙规则的设置 iptables -L在此说一下关于启动和关闭防火墙的命令:1..._linux防火墙服务找不到
C# DatrgridView表格控件的一些用法-程序员宅基地
文章浏览阅读109次。public class useDatrgrivView { string conn = null; string sqlComm = null; DataSet das = null; DataGridView GridView = null; //初始化,绑定 publ..._datrgridview 显示不了数据